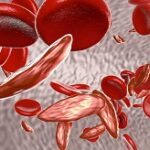

A talassemia é um tipo de anemia, em que ocorre uma má formação da fração “globina” da hemoglobina, resultando em redução de seus níveis sanguíneos. Essa má formação é provocada por uma falha genética, hereditária, com alteração do cromossomo 11 (talassemia beta) ou 16 (talassemia alfa), identificada a partir do exame do pezinho em recém-nascidos.
A pessoa com talassemia alfa pode ser portadora do gene e não manifestar a doença ou apresentar doença da hemoglobina H e hidropsia fetal. Na talassemia beta também pode ocorrer a presença do gene e não manifestação da doença (talassemia menor) ou o indivíduo pode desenvolver a doença (talassemia intermediária e talassemia maior).
Os sintomas são parecidos ao da anemia comum, como palidez, fraqueza e falta de ar. Porém, ao contrário da anemia carencial, na talassemia há um grande acúmulo de ferro, resultado do processo adaptativo do organismo que aumenta a absorção desse mineral na intenção de regularizar a síntese de hemoglobina. Por isso, seu diagnóstico envolve o exame do hemograma completo, dosagem de ferro sérico, ferritina, sobrecarga de ferro e eletroforese de hemoglobina. Já o tratamento inclui transfusões de sangue periódicas e uso de medicamento quelantes de ferro (que ajudam a tirar o excesso de ferro da circulação).


Nutricionista. Especialista em Nutrição Enteral e Parenteral pela BRASPEN. Especialização em Fitoterapia pela Ganep. Especialização em Nutrição Clínica pelo Centro Universitário São Camilo. Coordenadora de projetos e inovação do Ganep Educação e Nutritotal